
- Merck fights with New Zealand over coverage of its Keytruda cancer drug (statnews.com)Pembrolizumab (pharmac.govt.nz)

A row has broken out between Merck and New Zealand over a pricey new cancer drug in the latest example of how the cost of medicines is a flashpoint between drug makers and governments...At issue is Merck’s Keytruda (pembrolizumab), one of the new oncology treatments that harness the power of the body’s immune system to battle tumors. The medicine was approved in the United States two years ago to combat melanoma and, more recently, to tackle the most common form of lung cancer. Although priced at a hefty $150,000 a year, Keytruda is largely covered by public and private payers in the US...In New Zealand, the drug was approved last fall to treat melanoma. But since then, Pharmac, the government agency that decides whether coverage will be funded, has so far refused to endorse Keytruda. The agency contends evidence is lacking to verify whether the drug helps melanoma patients live longer compared with other new melanoma treatments or standard chemotherapy.
- One year after Zarxio approval, future of biosimilars remains unclear (modernhealthcare.com)Biosimilar drugs could save up to $110 billion by 2020: IMS (reuters.com)

A year ago, providers, plans and pharmacy benefit managers thought they were on the brink of a new era of competitive drug prices. The federal approval of the first biosimilar for sale in the U.S. was supposed to foster new products that offered big discounts on some of the most expensive treatments...But there's been no flood of new drugs and no lower prices since the Food and Drug Administration's approval of Sandoz's drug Zarxio...an alternative to Amgen's cancer therapy Neupogen, remains the only biosimilar for sale in the U.S...Since January, 59 biosimilars...have enrolled in the FDA's Biosimilar Product Development Program...the agency was actively seeking to recruit additional staff to meet the demand...The agency has been struggling with how to address interchangeability, or the ability to switch a patient onto a biosimilar drug from an original biologic, and vice versa, without impacting safety or efficacy...Naming conventions is another hot-button issue...FDA...proposed adding a four-letter suffix to the non-proprietary names shared with brand-name biologics...All of these issues raise questions as to whether the biosimilar market could ever reach its potential...Dan Mendelson, president of Avalere Health, said the biosimilar market will eventually be worth tens of billions, but he expected growth to occur slowly in the U.S., as consumers get more comfortable with choosing such drugs over their originator counterparts...
- Illicit drugs? How Brexit risks legal limbo for medicines (reuters.com)

A British vote to leave the European Union would threaten some prescription medicines with regulatory limbo, posing a legal headache for drugmakers, according to lawyers and industry officials...The highly regulated pharmaceutical sector has more at stake than most from a so-called Brexit, prompting top manufacturers GlaxoSmithKline and AstraZeneca, both of which oppose exit, to draw up detailed contingency plans...Currently, under EU rules, drugmakers launching a medicine get a single marketing approval that allows them to tap the entire European market of 500 million potential patients...In the event of a British exit, UK firms could no longer apply for or hold EU marketing authorizations, unless or until the UK negotiated to be part of the EEA. Licences would have to be transferred to businesses inside remaining member states..."The potential complexities around such issues as marketing authorization simply highlight the problems that could be faced by companies and patients alike in the event of exiting the EU,"...GlaxoSmithKline said leaving the EU would create uncertainty, add complexity and making some short-term disruption likely...
- FDA moves to increase competition among single-source generics (drugstorenews.com)

Based on the latest update to the Center for Drug Evaluation and Research’s Manual of Policies and Procedures, the Food and Drug Administration is looking to create more competition among generics — particularly for generics made by a single manufacturer... The updated MAPP outlines situations in which abbreviated new drug applications submitted by generics manufacturers will be eligible for an expedited review process, including submissions related to drug shortages, and legal requirements. Among them is the potential for expedited review for ANDAs related to what the agency calls "sole-source drugs" — drugs whose generic is manufactured by a single company...Submissions for drug products for which there is only approved product listed in the Prescription Drug Product List…of FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations...and for which there are no blocking patents or exclusivities may receive expedited review...
- China Eases Path for Foreign Drugmakers’ Hepatitis C Treatments (nasdaq.com)

China will grant four global drug companies priority-review status to launch groundbreaking new hepatitis C treatments in China, a rare move to open the lucrative market to foreign players...China's Food and Drug Administration expedites domestic drug applications to encourage innovation. But its lengthy drug-approval process for foreign companies means none of the direct-acting antiviral agents that have been shown to cure more than 90% of hepatitis C patients within a few months have been approved in China, which has among the highest rates of the disease in the world with an estimated 10 million people infected...It shows that the CFDA is serious about prioritizing important new innovative medicines that address real unmet medical need or improve substantially on what's currently available, whether they originate in domestic or [global] pharma companies...The policy also encourages foreign companies to manufacture drugs in China, saying companies will qualify for priority treatment if they submit applications for approvals in China simultaneously with U.S. and European Union approvals and use the same production standards as in those markets.
- Substances Doubtful for Bulk Drug Substances List Could Be INDs (ashp.org)Individual Patient Expanded Access Applications: Form FDA 3926 (fda.gov)

Pharmacists, physicians, and advocacy groups that want patients to use substances unlikely to be on the upcoming "bulk drug substances list" for compounders should consider submitting "treatment" investigational new drug applications, FDA personnel recently suggested...FDA-cleared treatment IND applications...offer a legal workaround that can benefit many patients...a compounding pharmacy, could submit a treatment IND, which once that was in place could be expanded to treat a large number of patients...ASHP stated, absent "significant" revision, FDA's current expanded-access IND application process will not facilitate access to any drug available only from compounders...Jarow (Jonathan Jarow, from the Center for Drug Evaluation and Research)...acknowledged that submission of an IND application has seemed difficult to many individual healthcare providers seeking a drug for a single patient..."There's now a special form in development that has not been finalized—it's available in draft form—that caters to that specific type of IND rather than the general form that's used for all types of INDs, which looks very complicated," he said...Form FDA 3926, also known as "Individual Patient Expanded Access Investigational New Drug Application," went on display in February 2015 as part of a draft guidance for the pharmaceutical industry....
- Welsh gov overrules NICE rejection of Celgene’s pancreatic cancer drug (pharmatimes.com)

Patients with pancreatic cancer living in Wales will no doubt welcome news that Celgene’s Abraxane will continue to be available on the National Health Service in the country, despite a decision by the National Institute for Health and Care Excellence to reject its use...NICE confirmed back in November that Celgene’s Abraxane (nab-paclitaxel) will not be routinely funded for use in combination with gemcitabine for treating pancreatic cancer on the NHS in England and Wales, claiming that the drug’s cost does not justify its benefit to patients...the Institute said while the Abraxane/gemcitabine combination was more effective than the latter drug alone, it resulted in more serious side effects, and also caused more adverse events than a gemcitabine/capecitabine regimen despite having similar effectiveness...in a rare move, the Welsh government (All Wales Medicines Strategy Group) has now stepped in to ensure continued access for metastatic pancreatic cancer patients in Wales, on the back of data from the MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) study, which showed an increase in median overall survival of 1.8 months with Abraxane in combination with gemcitabine, when compared to gemcitabine alone.
- FDA Review Times Steadily Decreasing, Report Finds (raps.org)
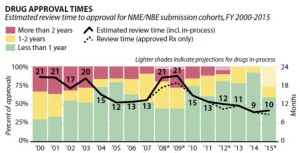
Despite wide variations across therapeutic areas, FDA review times for new drugs have steadily declined since 2009, according to a report released...by the California Life Sciences Association and Boston Consulting Group...Back in 2009, FDA was averaging 21 months for reviewing new molecular and biologic entities, but five years later, that average has been cut by more than half to nine months in 2014. In addition, the number of applicants waiting more than two years for an FDA review has fallen from more than 25% of applicants in 2000 to none by 2013.
- Sanofi science chief on Zika: It’s time to disrupt traditional vaccine development (fiercevaccines.com)

The World Health Organization has warned that the Zika R&D frenzy may not culminate in a vaccine in time for the current outbreak, but Sanofi Chief Scientific Officer Gary Nabel won't take that for an answer. Nabel says it can be done, but it means turning the traditional vaccine development model on its head...In an interview...Nabel highlighted that the classic response to emerging and infectious diseases, such as MERS and Ebola, has been inadequate...We just run from one crisis to another…That's no way to protect the world's population...The traditional vaccine development timeline typically takes 5 years or more to produce a marketable vaccine... To have a chance at accelerating this for emerging diseases, biotechs and pharmas, regulators and government agencies need to come together and unpack this model, identifying where time can be saved and exploring different ways to go about clinical trials...(Nabel) identified WHO as "symptomatic of the problem." While it has "the best of intentions," it doesn't have the wherewithal to follow through...The agency has declared Zika a global public health emergency and in February called for $56 million in funding to combat the virus. Just over a month later, only $3 million--5%--in funding has trickled in...
- FDA changes policy to prevent the next Martin Shkreli (statnews.com)

...the Food and Drug Administration...made a policy change that may prevent companies from pulling a Martin Shkreli...The agency plans to expedite reviews of applications for generic drugs where only one treatment is currently sold. The shift was prompted by public outrage that erupted last fall when Turing Pharmaceuticals...bought a life-saving drug called Daraprim and promptly jacked up the price by 5,000 percent...We identified a gap and were able to identify a path forward...The change being made (allows the agency) to capture circumstances when the only approved product on the market is a generic drug...Even though Turing does not hold a patent on the medicine...company was able to increase the price as it did because there was no generic competition. The drug maker runs a closed distribution system, and as a result, Turing has a monopoly on Daraprim...This should provide a faster way to inject competition in the marketplace, so that the price gougers can’t get away with what they’re doing...the policy change is retroactive, which means the agency will review pending applications to see if any merit an expedited review.