
- FDA Guidance on Biosimilar Substitution Needs Work, Commenters Say (bna.com)

The FDA needs to clarify its standards for showing a biosimilar drug is interchangeable with the original biologic and how the biosimilar should be named and labeled, commenters said in response to an agency draft guidance...The Food and Drug Administration draft...described how an applicant can establish that a biosimilar, a highly similar, less expensive version of an FDA-approved biologic drug, can be substituted for the biologic on which it is based without a physician’s approval…If you’re a biosimilar applicant, you’re happy the FDA has provided the guidance, but you think it’s asking for too much information...If you’re the owner of the patent for the original biologic, you strongly support the need for the switching studies the FDA requires in order to show that switching from the original biologic to the biosimilar is safe. If you’re an organization that represents patients, you want assurances the interchangeable biosimilar is safe and effective for each condition for which the original biologic is approved...
- No scandal here, say rare disease drugmakers, as pricing scrutiny lands on their doorstep (fiercepharma.com)

Rare disease drugmakers get that their typically high prices are under the lens these days—just like everyone else’s. But they don’t all necessarily agree that the scrutiny is fair...Shire CEO Flemming Ornskov...understands why the field has landed in the pricing spotlight, he doesn’t think it belongs there…list prices that would have flown just a couple years back have become unacceptable in the public eye. Rare-disease drugmakers’ go-to pricing justifications—that their meds are innovative and used by few patients, meaning they're not too big a hit on payers—are no longer getting it done...Gamida Cell...thinks Nicord—its candidate stem cell treatment for blood cancer patients—can "support a justification for a higher price,"…it’s the med’s cost-saving potential that will ultimately "dictate the pricing,"...
- The 340B Program Hits $16.2 Billion in 2016; Now 5% of U.S. Drug Market (drugchannels.net)

The 340B Drug Pricing Program’s explosive growth continues...discounted sales hit $16.2 billion in 2016. That’s a 34% increase over the 2015 figure. Consequently, the 340B program accounted for 5.0% of the total U.S. drug market in 2016...Covered entities are generating billions in untraceable profits from this fast-growing program. Hospitals, which make up the vast majority of 340B purchases, should be required to account clearly for the billions the program provides them...Some hospitals have published high-level descriptions of how they spend 340B funds, but none of the articles fully accounts for the billions of dollars in 340B profits. Much more transparency is required, because the dollars are very large and growing very quickly…Given the program's rapid growth and its channel distortions, Congress urgently needs to refocus the program on genuine safety-net providers and financially needy patients.
- Drugs approved with limited data aren’t always well-tested later (reuters.com)

New medicines that win U.S. marketing approval without conclusive evidence of their effectiveness aren't always proven to work after they go on sale, a recent research review suggests...Researchers focused on medicines approved for sale based on single pivotal trials or based on what's known as "surrogate markers," such as lab tests and signs of risk for disease such as cholesterol levels instead of true clinical outcomes like heart attacks or deaths. Many times no follow-up studies were published after these medicines went on sale, and when studies were published they often continued to rely on surrogate markers to suggest potential effectiveness...speeding up the approval process increases our collective uncertainty about drugs' benefits and safety...This exposes patients to risks - the risk that they are spending their resources on therapies that do not work as well as expected as well as the possibility that they are taking drugs that have underlying safety risks that have not yet been figured out… researchers examined published studies of 117 medicines approved for treating 123 medical conditions by the U.S. Food and Drug Administration...based on either a single pivotal trial or on trials that relied on surrogate endpoints...no follow-up studies were published for 43 of the 123 approved indications, or 35 percent...
- FDA ad police smack Orexigen for leaving black-box risk out of Contrave TV ad (fiercepharma.com)

It’s taken five months, but the FDA has finally issued its first untitled letter of the year for promotional infractions. The agency's promotional watchdogs scolded Orexigen Therapeutics for alleged infractions in a TV commercial for weight loss drug Contrave...the Office of Prescription Drug Promotion says the television ad misleads viewers by touting Contrave's benefits and downplaying its risks. For instance, the letter says the spot omits important risk information—including potential neuropsychiatric reactions, such as suicidal thoughts, that are highlighted in a black-box label warning...By omitting serious risks associated with Contrave, the TV ad misleadingly suggests that Contrave is safer than has been demonstrated...Orexigen is currently addressing the OPDP guidance...
- Nevada Senate Passes Insulin-Price Bill Tough on Drugmakers (lasvegas.cbslocal.com)
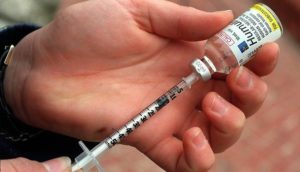
A bid in Nevada to force America’s three insulin manufacturers to turn over information on the...prices of...hormone treatment for people with diabetes advanced with bipartisan support...The Democratic (Democrat) proposal has transitioned from a first-of-its-kind price control to a measure focused on pricing transparency...it retains what would be some of the toughest regulations on pharmaceutical companies in the nation...It would require drugmakers to annually publish the list prices they set and profits they make on insulin, as well as the total amount of insulin discounts they give market middlemen — specific data points currently largely kept confidential...Some pharmaceutical companies, including insulin-maker Sanofi SA, have voluntarily released recent data on their price increases...Market experts say transparency alone won’t lower patient costs...
- Study: One-third of docs trust pharma content on HCP sites (mmm-online.com)

Only 34% of physicians find pharmaceutical content on HCP (healthcare professional)sites to be trustworthy, according to a new study by Decision Resources Group's Manhattan Research...We found that pharma is missing the mark when it comes to providing online content that physicians need in an easy-to-use way...DRG...surveyed 2,784 U.S. physicians in more than 26 specialties and found that only 27% of them viewed pharmaceutical websites as credible sources of professional information, compared to 77% of them who viewed third-party HCP-facing websites credible...62% of the physicians surveyed said that the information they get from drugmakers on third-party sites such as HCP sites and online journals “are always ads.”...What physicians need are resources for patient education, scientific information on drugs to help them make better treatment decisions, and continuing medical education, said Arnold. Seventy percent of physicians said it's crucial for drugmakers to provide educational resources rooted in science to gain their trust, yet half of them agree that no drugmakers are providing quality scientific online...
- Accelerated FDA approval and pricey drugs make a rotten combo, doctors argue (fiercepharma.com)

Should drugs that win FDA approval on an accelerated path—often without strong evidence of efficacy—command the same high prices as products that undergo the full menu of agency scrutiny?...Not necessarily, two doctors contend in a new journal article. And they have some ideas for bringing the costs of those drugs way down, at least until their makers can prove they really work...Accelerated approval is a problem because it forces Medicare and Medicaid to pay for drugs that can cost hundreds of thousands of dollars and yet may not work for many patients...private insurers may decline to cover those drugs, denying patients access to products that were supposed to get to them faster...The FDA requires drugmakers who gain accelerated approval to perform post-marketing studies to prove efficacy—and therein lies an opportunity for the healthcare system to save money...(authors) propose. They suggest drug manufacturers be required to discount products until confirmatory trials are complete, or that a portion of the full price they’re charging be held in escrow until the results of the trials are available...We see the value in getting products that qualify for accelerated approval to market for patients, but we believe the price paid by taxpayers should reflect the strength of the available evidence about the drug’s clinical impact...
- European countries offer goodies in bid to win EMA after Brexit (fiercepharma.com)

Even before European officials present the groundwork for relocating the bloc’s drug regulator, a fight has emerged between countries vying for the economic lift and status that come with hosting the authority, using perks like child care as ammo...Some 20 countries in Europe hope to host the European Medicines Agency after the U.K.’s vote to leave the union last summer...And even though the EU hasn’t announced its official criteria for the move, the countries aren’t hesitating to make their case by showcasing local lifestyle perks and more...Countries in Europe are offering language lessons, local scenery and child care as they push to win the EMA, according to the news service. They’re seeking to attract an agency that could bring an economic lift worth an estimated €1 billion and employs about 900 experts.
- HHS Secretary Pushes to Cut FDA Appropriations, Replace With More Industry Fees (raps.org)

Secretary of Health and Human Services...Tom Price is continuing to push the Senate to further increase the industry fees paid to the US Food and Drug Administration...which would upend the agreed-to amounts negotiated by FDA and industry for the next five years, and allow for further cuts to the agency’s congressional appropriations...the Senate Health, Education, Labor & Pensions committee advanced a bill reauthorizing the user fee programs for prescription drug, medical device, generic drug and biosimilar industries without the additional fees that President Donald Trump requested in his budget blueprint for the next fiscal year...The fees included in the bill were what both sides negotiated for PDUFA VI, MDUFA IV, GDUFA II and BsUFA II...Price wrote in a letter...to Sen. Patty Murray, the ranking member of the HELP committee: "To ensure the FDA has the critical resources needed to keep pace with this field, the President's Budget Blueprint proposes to increase and restructure the medical product user fee programs at FDA to be 100 percent user fee supported programs, with no funding triggers that require budget authority financing."...