
- Woodcock to step up to interim FDA chief as she and Sharfstein are vetted for permanent job: reports (fiercepharma.com)

Janet Woodcock
FDA veteran Janet Woodcock will take over as commissioner, at least temporarily, according to media reports, and former deputy FDA chief Joshua Sharfstein is at the top of the list for the permanent job...President-elect Joseph Biden has tapped Woodcock as interim FDA commissioner while his team searches for a permanent replacement...Woodcock now oversees COVID-19 therapy development as part of Operation Warp Speed and has headed up the Center for Drug Evaluation and Research since 2008...READ MORE
- Will Pharma Meet the Drug Tracking Deadline? (biopharminternational.com)

Manufacturers and trading partners struggle to meet drug tracking requirements...As FDA and industry near the halfway mark in the 10-year process for establishing a national electronic drug tracking system by 2023, there’s considerable concern among pharma companies, wholesaler/distributors, and pharmacists about meeting the deadline. The process for establishing the rules and infrastructure for the track-and-trace system envisioned in the Drug Supply Chain Security Act, part of the Drug Quality and Security Act of 2013, is proving to be complex and challenging for all parties...FDA recently delayed requiring drug manufacturers to imprint unique product identifiers on individual packages by November of 2017, saying it would not enforce that policy until Nov. 27, 2018. While major pharma companies are meeting the earlier time-frame for serializing and identifying drug packages, many generic-drug makers and contract manufacturers reported confusion over who is responsible for devising the identifiers and for confirming compliance with requirements. And while manufacturers applauded gaining an additional year to fully identify individual drug packages, pharmacists and other supply chain partners raised concerns that the delay would make it even more difficult for them to comply with reporting requirements for 2018 and 2019...
- FDA Says Real-World Evidence Could Generate ‘Incorrect or Unreliable Conclusions’ (raps.org)

...top Food and Drug Administration officials published an article in the New England Journal of Medicine (Real-World Evidence — What Is It and What Can It Tell Us? )...calling into question some of the potential uses of real-world evidence but also acknowledging that real world research and the concepts of a planned intervention and randomization “are entirely compatible."...while acknowledging that such (real-world) evidence “can inform therapeutic development, outcomes research, patient care, research on health care systems, quality improvement, safety surveillance, and well-controlled effectiveness studies,” the authors caution that “the confluence of large data sets of uncertain quality and provenance, the facile analytic tools that can be used by nonexperts, and a shortage of researchers with adequate methodologic savvy could result in poorly conceived study and analytic designs that generate incorrect or unreliable conclusions...“Accordingly, if we are to realize the full promise of such evidence, we must be clear about what it is and how it can be used most effectively, and we must have appropriate expectations about what it can tell us,”...
- 2015: Another Strong Year for Patients in Need of New Drug Therapies (blogs.fda.gov)Novel New Drugs Summary 2015 (fda.gov)
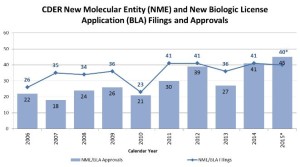 I’m (John Jenkins,Director of the Office of New Drugs) pleased to report another strong year for FDA approvals of novel new drugs, which offer many patients new treatment options for serious and life-threatening conditions. In 2015, FDA’s Center for Drug Evaluation and Research approved 45 novel new therapies – significantly more than the average of 28 we have approved during the previous nine years of this decade...During this past year, we approved many new drugs to treat various forms of cancer, including four to treat multiple myeloma, and others to treat lung, skin, breast, brain, colorectal, and other cancers. We also approved new drugs to treat heart failure, high cholesterol, cystic fibrosis, and irritable bowel syndrome, as well as the first approved reversal agent for a commonly-used blood thinner...Here are a few highlights of these approvals:
I’m (John Jenkins,Director of the Office of New Drugs) pleased to report another strong year for FDA approvals of novel new drugs, which offer many patients new treatment options for serious and life-threatening conditions. In 2015, FDA’s Center for Drug Evaluation and Research approved 45 novel new therapies – significantly more than the average of 28 we have approved during the previous nine years of this decade...During this past year, we approved many new drugs to treat various forms of cancer, including four to treat multiple myeloma, and others to treat lung, skin, breast, brain, colorectal, and other cancers. We also approved new drugs to treat heart failure, high cholesterol, cystic fibrosis, and irritable bowel syndrome, as well as the first approved reversal agent for a commonly-used blood thinner...Here are a few highlights of these approvals:- More than one-third of the novel new drugs CDER approved in 2015 were identified by FDA as “first-in-class,” for example, drugs that use a new and unique mechanism of action for treating a medical condition;
- More than 40% of these new therapies were approved to treat rare or “orphan” diseases that affect 200,000 or fewer Americans–Americans who often have few or no drug treatment options;
- 60% of CDER’s novel new approvals for 2015 were designated in one or more categories of Fast Track, Breakthrough, Priority Review, or Accelerated Approval. Each of these designations helps speed the development and/or approval process and is designed to help bring important medications to the market as quickly as possible; and
- 64% of CDER’s novel new approvals were approved first in the United States before any other country.
- Gottlieb Proposes Modernization of Drug Review Office (biopharminternational.com)

...FDA Commissioner Scott Gottlieb, MD, announced that the Center for Drug Evaluation and Research would be taking steps to modernize the organization and functions of the Office of New Drugs in order to address scientific and medical advances within the industry. The goal is to make the review process more integrated across science and regulatory expertise. Janet Woodcock, director of CDER, plans on elevating the role of FDA scientists and medical officers and providing these officers with more tools and support “to advance the clinical and regulatory principles that the FDA uses to evaluate new drugs for safety and efficacy.”...Other changes will include the development of guidance documents, giving review staff more time with sponsors, and getting sponsors involved earlier in the development process. Engaging disease specialists, academic researchers, regulatory partners at other agencies, and patient groups is also a goal of CDER.
- FDA Details Plans for More Efficient Inspections, Facility Evaluations

The US Food and Drug Administration's Center for Drug Evaluation and Research and Office of Regulatory Affairs will soon launch an effort to streamline the two offices' inspection and facility evaluation efforts...CDER Director Janet Woodcock and Associate Commissioner for Regulatory Affairs Melinda Plaisier said it is vital that the two offices quickly implement the plan in order to meet commitments under the recently reauthorized user fee agreements, specifically citing the agency's promise to communicate final inspection classifications to generic drugmakers within 90 days of an inspection beginning in October 2018...We plan to operationalize the plan in the fall of 2017 for nearly all human drugs...FDA details the plan—which includes specific operating models for pre- and post-approval inspections, surveillance inspections and for-cause inspections—in a 20-page white paper obtained by Focus entitled Integration of FDA Facility Evaluation and Inspection Program for Human Drugs: A Concept of Operations...
- FDA moves to increase competition among single-source generics (drugstorenews.com)

Based on the latest update to the Center for Drug Evaluation and Research’s Manual of Policies and Procedures, the Food and Drug Administration is looking to create more competition among generics — particularly for generics made by a single manufacturer... The updated MAPP outlines situations in which abbreviated new drug applications submitted by generics manufacturers will be eligible for an expedited review process, including submissions related to drug shortages, and legal requirements. Among them is the potential for expedited review for ANDAs related to what the agency calls "sole-source drugs" — drugs whose generic is manufactured by a single company...Submissions for drug products for which there is only approved product listed in the Prescription Drug Product List…of FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations...and for which there are no blocking patents or exclusivities may receive expedited review...
- CDER Publishes Drug Safety Report – FDA’s Center for Drug Evaluation and Research has published its second annual report on key safety programs and activities. (biopharminternational.com)

...FDA’s Center for Drug Evaluation and Research released its second annual Drug Safety Priorities report, which details drug safety initiatives carried out by CDER and FDA. The report highlights drug safety program milestones and gives an update on goals achieved in 2017. Efforts by FDA to ensure drug safety science, surveillance, and oversight are discussed...Detailed in the report are the agency’s efforts on pharmacovigilance, medication errors, and risk management...The report also goes into detail about the agency’s views on how real-world evidence can advance drug safety. An update on the agency’s efforts to combat the opioid crises is also provided...
- New FDA Database Ensures Timely Access to Drug Safety Labeling Changes (aafp.org)

...a new FDA database that drastically improves timely access to information on drug safety labeling changes...part of the FDA's official website, was launched a few months ago by the FDA's Center for Drug Evaluation and Research...The agency now is leading a push to...get...critical safety data...much more quickly than in the past...The user-friendly and searchable database (www.accessdata.fda.gov) provides updates on labeling information, usually within days of safety labeling changes…Safety labeling changes have been available online...for years, but the information was posted on a monthly basis. That meant when a labeling change was approved early in a month, the information was not made public until the following month -- weeks later...
- Toward saner drug pricing (washingtontimes.com)
If the FDA acts more quickly, lower costs and better patient outcome will follow...In a January hearing, Sen. Hatch queried Dr. Janet Woodcock, director of the Food and Drug Administrations’s Center for Drug Evaluation and Research, whether the agency’s backlog of some 3,500 lower-cost generic drugs awaiting approval was contributing to the cost consumers pay for drugs...Dr. Woodcock conceded that...slow-paced drug approvals contribute to high costs...Eighty-eight percent of all prescriptions in the U.S. are generics. If the FDA acted more quickly with both generics and new drugs, overall health costs would go down and patient health outcomes would improve...By quickly approving generics, lower cost versions would be available without unfairly infringing on profits (and thus stifling innovation) that pharmaceutical companies need to recoup the high costs to bring new drugs to market...Ms. Warren (Sen. Warren, Mass, Democrat) said...She’d like us to believe that Mr. Shkreli is representative of the broader industry and only government price controls can protect consumers from what appear to be an ever-rising cost of drugs...Rep. Stephen Lynch, Mass. Democrat, proposed what he amusingly called a "poison-pill amendment," which would allow Congress to decide when drug prices are too high, and then just eliminate patent exclusivity and "contract with DARPA — our government research labs — to produce your drugs at no cost to the consumer. That’s what we can do."...The Lynch and Warren approach will do one thing for sure. It will have a chilling effect on much needed private-sector investment in pharmaceutical research...the "market-failure, price control" argument would translate to higher overall treatment costs...