
- FDA approves first generic of Mylan’s EpiPen (pharmaceutical-technology.com)

The Food and Drug Administration has approved the first generic version of the EpiPen and EpiPen Jr auto-injector for severe allergic attacks, including life-threatening anaphylaxis....The EpiPen was approved in 2007 and is marketed by Mylan. However, since then both the company and its product have been involved in a number of scandals. Notably the price of the EpiPen has increased from $57 in 2007 to approximately $600 in 2016 and in May, the FDA had to place Mylan’s EpiPen on its official shortages list as a result of manufacturing delays...The newly approved generic epinephrine auto-injector will be available in 0.3mg and 0.15mg strengths and is marketed by Teva. This the company’s second attempt to get its generic approved by the FDA; it was rejected in 2016...FDA commissioner Scott Gottlieb said: “Today’s approval of the first generic version of the most-widely prescribed epinephrine auto-injector in the US is part of our longstanding commitment to advance access to lower cost, safe and effective generic alternatives once patents and other exclusivities no longer prevent approval.
- China drug scandals highlight risks to global supply chain (cnbc.com)

The drug safety scandals...have underlined the risks to international consumers posed by weak oversight in China, the world's largest supplier of active pharmaceutical ingredients...The European Medicines Agency and the US Food and Drug Administration issued alerts over a cancer-causing ingredient used in a blood pressure medication, supplied by Chinese company Zhejiang Huahai, resulting in a recall of affected drugs...Then Beijing announced that hundreds of thousands of substandard vaccine doses had been sold in China, prompting a public outcry. Senior executives were arrested at the pharma company, Changsheng Biotech, which was also accused by authorities of forging data during the production of rabies vaccines...China is home to thousands of API producers, with exports worth $29bn last year...its producers supply ingredients for generic drugmakers such as Teva Pharmaceutical and multinationals including Johnson & Johnson and Novartis. About 80 percent of APIs used in the US come from China and India…Warnings to Chinese companies published by the FDA and EMA in recent years show that dozens have violated standards, mainly relating to record-keeping during the manufacturing process. In several cases the exporters shipped large volumes of product before the infractions were discovered...
- Blood Pressure Medicine Is Recalled (nytimes.com)

The Food and Drug Administration has announced a voluntary recall of a widely prescribed blood pressure medication made in China, reviving fears about the safety of imported drugs...Three companies that sell the generic drug, valsartan, in the United States agreed to recall it after the F.D.A. said it might be tainted by N-nitrosodimethylamine (NDMA), considered a probable human carcinogen. The agency is still investigating, but said the contamination was believed to be related to changes in the way that valsartan was manufactured...All of the valsartan that is being recalled was made in China by the same company, Zhejiang Huahai Pharmaceutical Co. Ltd. It is distributed in the United States by three companies: Major Pharmaceuticals; Teva Pharmaceutical Industries, Ltd.; and Solco Healthcare... Other companies that market the drug, not subject to the recall, are Sun Pharma, Mylan, Jubiliant, Aurobindo and Hetero...The safety of imported drugs has long been debated. The F.D.A. said it would continue to investigate the levels of NDMA in the recalled products, determine the possible effect on patients who have been taking them, and assess what measures can be taken to reduce or eliminate the impurity from future batches...
- EMA Recommends First CART-T Cell Therapies (biopharminternational.com)

...the European Medicines Agency recommended Novartis’ Kymriah (tisagenlecleucel) and Kite Pharma’s Yescarta (axicabtagene ciloleucel), chimeric antigen receptor (CAR) T-cells therapies for blood cancer, for approval in the European Union...Kymriah and Yescarta are the first CAR T-cell treatments to be recommended by the agency. In August 2017, Kymriah became the first CAR-T therapy approved by FDA in the United States, with Yescarta becoming the second in October 2017...Both drugs are also the first treatments supported through EMA’s Priority Medicines scheme to receive positive opinions from the Committee for Medicinal Products for Human Use...
- FDA OKs first generic under new approval pathway (drugstorenews.com)

This new approval pathway was created to expedite the development and review of a generic drug for products that lack competition...The FDA gave the nod for Apotex’s potassium chloride oral solution...“Today’s approval marks the successful implementation of a new program designed to encourage generic drug development for products with inadequate generic competition,” said FDA commissioner Scott Gottlieb, in a press statement...“The quick implementation of this new pathway is part of our broader effort to foster generic competition and help address the high cost of drugs. So are our efforts to narrow the time it takes for generic drugs to reach the market by reducing the number of review cycles that generic applications typically undergo. This new generic drug application was also approved in its first cycle of review. This approval demonstrates that the competitive generic therapy pathway is efficient and open for business. This pathway is a key step in making safe and effective generic drugs available to patients quickly and ensuring there’s adequate competition so patients have affordable access to the treatments they need,” said Gottlieb.
- FDA seals loophole that allowed some drugmakers to avoid pediatric clinical trials (medcitynews.com)

PREA loophole had allowed companies to receive orphan drug designations for diseases common in adults...The Food and Drug Administration moved...to close a loophole that had inadvertently excused some drugmakers from having to conduct clinical trials in children for drugs to treat diseases that commonly affect adults...the FDA announced that it had finalized a draft guidance...intended to close a loophole in the Pediatric Research Equity Act that allowed drug companies making medicines for non-orphan diseases in adults – meaning those affecting more than 200,000 patients – to get pediatric subpopulation designation. Consequently, companies were exempt from conducting the clinical trials in children that PREA would normally have required...“Addressing the inadequate testing of drugs in pediatric populations has been a priority for the FDA, the medical community and Congress and has led to important laws to ensure this important, vulnerable population is not overlooked,” FDA Commissioner Scott Gottlieb said...
- FDA looks at incentives, manufacturing ‘interventions’ to solve drug shortages (fiercepharma.com)

The FDA will consider creating a list of “essential drugs” and financial incentives to drugmakers that manufacture them as it continues to grapple with the drug shortages that plague U.S. hospitals and caregivers...FDA Commissioner Scott Gottlieb, M.D...announced the formation of a new Drug Shortages Task Force and named Keagan Lenihan, the FDA’s associate commissioner for strategic initiatives to lead it...“I’m charging the shortages task force to delve more deeply into the reasons why some shortages remain a persistent challenge,” Gottlieb said. “The charge to this new task force is to look for holistic solutions to addressing the underlying causes for these shortages.”...The FDA is already taking steps to support new technologies that can improve manufacturing and help reduce the chance that supply disruptions will occur...the FDA may want to consider “more significant interventions” than it currently employs. “We want to make sure we aren’t discouraging investment for manufacturing drugs that are more likely to go into shortage, and thus working against our own goals." One suggestion is to grant the FDA authority to require applicants of certain drugs to conduct a “risk assessment to identify the vulnerabilities in their drug supply that could cause a shortage," and to establish risk mitigation plans in advance to address those weaknesses...
- FDA Anticipates Need for Expanding National Drug Code, Calls for Industry Input (raps.org)
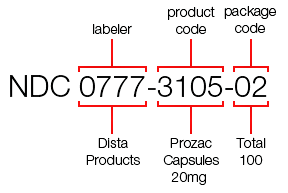
The Food and Drug Administration announced...it plans to hold a public discussion on expanding the National Drug Code format and the impact on industry...FDA currently assigns 10-digit NDCs for the purposes of identifying drugs in the country and the format is comprised of three codes regarding the labeler, the product and the package...“FDA anticipates that it will run out of 5-digit labeler codes in approximately 15 years,” the agency wrote in the notice on the 5 November public hearing. “FDA will begin assigning 6-digit labeler codes at some point in the future due to exhaustion of 5-digit labeler codes.”...Prior to implementing the new formats for longer configurations, FDA officials will seek feedback from meeting participants on the potential impact on companies’ bottom line and drug safety, as well as “issues associated with the current lack of NDC uniformity in the marketplace.”
- EU drug regulators step up work to prepare for ‘no deal’ Brexit (reuters.com)

Drug regulators across Europe are hiring extra staff and increasing their workload as the role of British experts in the EU-wide system of medicines supervision winds down ahead of Brexit...Britain has already stopped taking on new projects that will extend beyond March 29, 2019 and is preparing to hand over existing drug review work to other countries...Despite a vote by UK members of parliament this week calling for Britain’s continued participation in the Europe regulatory network for medicines, there is no certainty that any such deal will be reached...That reflects the wider lack of clarity over Britain’s future relationship with the world’s biggest trading bloc after it leaves the EU next March...The Brexit-induced disruption also comes at a time when regulators are having to grapple with oversight of a range of new health technologies, such as gene therapy, and a slew of big data on health outcomes...Global drug companies, including UK-based GlaxoSmithKline and AstraZeneca, have been vocal in calling for continued close EU-UK ties after Brexit. The issue is also important to many Japanese drugmakers that have made Britain their European base.
- FDA to Consumers: Stay Away from Maximum Powerful (ptcommunity.com)

The Food and Drug Administration is advising consumers not to purchase or use Maximum Powerful, a product promoted for sexual enhancement that contains sildenafil, the active ingredient in Viagra. This product was identified during an examination of international mail shipments...The announcement is part of the agency’s effort to inform the public of a growing trend of dietary supplements or conventional foods with hidden drugs and chemicals. The culprit products are typically promoted for sexual enhancement, weight loss, and bodybuilding and are often represented as being “all natural.”...the FDA noted that it is unable to test and identify all products marketed as dietary supplements that have potentially harmful hidden ingredients. As a result, the agency issued a blanket warning that consumers should exercise caution before purchasing any product touted as improving sexual enhancement, helping with weight loss, or building up muscle...