
- Manufacturers Seek Strategies for Ensuring Quality of Innovative Therapies (biopharminternational.com)

Continued industry investment in advanced biologics will raise further regulatory challenges and require innovative manufacturing systems...The challenges in producing cellular and gene therapies and other cutting-edge products that meet standards for safety, efficacy, and quality was high on the agenda for biopharmaceutical companies at PDA/FDA annual joint regulatory conference...
- Center for Biologics Evaluation and Research...is expediting the development and approval of human cellular and tissue products...vetting new regenerative medicine advanced therapies, including those utilizing gene-editing technology to treat malignancies and diseases...
- National Institutes of Health...cell biology and tissue engineering to advance regenerative medicine...these products require very different manufacturing and dosing processes and raise multiple challenges for scale-up and for keeping cells alive during processing...
- CBER’s Office of Tissues and Advanced Therapies...devising standards for characterization and potency assays for regenerative products, stem cell therapies, therapeutic vaccines, gene therapies, antivenins and certain combination products…importance of controlling the manufacturing process for these products and...challenges in achieving the right level of product characterization based on critical quality attributes.
- Gottlieb vows to shake up the FDA, backing a trend toward faster drug development (endpts.com)

FDA commissioner Scott Gottlieb has sounded a crystal clear warning over the high — and growing — cost of drug development. And in a speech to regulatory execs...Gottlieb committed the FDA to backing up more efficient drug development programs with new measures to clear the regulatory path for developers barreling ahead to relatively swift pivotal data in search of an accelerated OK...Gottlieb started by outlining a bleak picture in drug R&D, noting that the economic model for drug development is broken. It costs too much to develop a drug so it can be approved for marketing. And costs are swelling fast at the discovery end of the business, which will help swamp a system that already doesn’t work particularly well...Gottlieb held up some of the rapid-fire clinical trials we’ve been seeing in the cancer field as a model for what can work, paving the way to the accelerated approval pathway at the FDA. And he believes...that moving drug development into the fast lane can reduce R&D costs and thereby allow biopharma companies to pass on savings to patients through lower costs.
- Feds seize smallpox vaccine from clinic injecting it into cancer patients (theverge.com)

The US Food and Drug Administration has stopped a California company from continuing to inject the smallpox vaccine into the tumors of cancer patients...US marshals seized five vials of the smallpox vaccine from San Diego-based StemImmune Inc, which was using them as part of an unproven method for treating tumors...he vaccine is not commercially available and is only reserved for people who are at very high risk for developing smallpox, so it’s not clear how StemImmune got the vials to begin with. Luckily, the vaccine is not made from the actual smallpox virus and cannot give anyone the disease...It was...not meant to be used the way StemImmune was doing so, which was by mixing it with stem cells that come from body fat and injecting it into the tumors of patients at California Stem Cell Treatment Centers in Beverly Hills and Rancho Mirage...The FDA is encouraging consumers who have tried the treatment and had bad effects to use its MedWatch Adverse Event Reporting program.
- FDA panel: Not enough data to OK “abuse-deterrent” opioid (kolotv.com)

Food and Drug Administration advisers voted against approving a new opioid painkiller with a unique feature for deterring abuse: It releases a deep-blue dye if someone tries to get high by crushing, chewing or snorting pills...Panelists voted overwhelmingly against approving Intellipharmaceutics International's generic version of extended-release Oxycontin, a key drug in the U.S. opioid addiction epidemic. The FDA usually follows its advisers' advice...Doctors and scientists raised many concerns about the dye's safety for intended patients, and its effectiveness in deterring opioid abusers. Most said the company hadn't done enough studies of the drug...Some suggested blue stains around the mouth or nose from trying to abuse the drug could become popular among addicts. Others said the dye's "Scarlet Letter" shaming approach was insensitive...also...the company hadn't provided enough data to show that addicts couldn't find a way to convert the pills' active ingredient, oxycodone, into a liquid that could be injected...Advisers noted that the company hadn't studied issues such as whether the nasal irritant in the pill would prevent abusers from trying to snort it after crushing and whether the blood dye would discourage abuse attempts.
- Exclusive: FDA plans new compounding pharmacy policy, agency head says (reuters.com)

The head of the U.S. Food and Drug Administration said...the agency is working on a new policy that would encourage more compounding pharmacies to register under...the Drug Quality and Security Act, which aimed to bring more compounding pharmacies...under the authority of the FDA rather than state pharmacy boards...The law created a category of “outsourcing facilities” that could register with the FDA, allowing them to sell products in bulk to hospitals and physician practices without prescriptions for individual patients...In exchange, those compounders would have to follow federal manufacturing standards and subject themselves to routine inspections...around 70 firms have registered as outsourcing facilities...compounders that did not register with the FDA would remain under state oversight, and...could only compound drugs based on prescriptions for specific patients...Gottlieb said that in order to encourage more compounders to register, the FDA would release draft guidance in the next two months reflecting its intention to adjust its enforcement priorities based on the size of registered compounders and the riskiness of their products...We’re looking at ways we can provide more of a gradation in our regulatory architecture so we don’t have a one-size-fits-all approach...
- F.D.A. Accuses EpiPen Maker of Failing to Investigate Malfunctions (nytimes.com)

The Food and Drug Administration...accused the drugmaker Pfizer of failing to properly investigate reports of malfunctioning EpiPens, including incidents when patients died or became severely ill after the device failed to work. Pfizer manufactures the EpiPen, which treats allergic reactions, for the drugmaker Mylan...In a warning letter...the agency said Meridian Medical Technologies, which is a unit of Pfizer, did not adequately look into problems with a crucial component of the EpiPen — the mechanism on the device that insures that it fires and delivers the proper dose of epinephrine, which stops an allergic reaction... the company failed to conduct a proper investigation even though it received numerous complaints about problems with activating the device. "Our own data show that you received hundreds of complaints that your EpiPen products failed to operate during life-threatening emergencies, including some situations in which patients subsequently died...
- Libertarian billionaire Peter Thiel funds “unethical” offshore human test of herpes vaccine, skirting FDA rules (salon.com)St. Kitts Launches Probe Of Herpes Vaccine Tests On U.S. Patients (khn.org)
Defying U.S. safety protections for human trials, an American university and a group of wealthy libertarians...are backing the offshore testing of an experimental herpes vaccine...Peter Thiel, invested $7 million in the ongoing vaccine research...Southern Illinois University also trumpeted the research and the study’s lead researcher...Neither the Food and Drug Administration nor a safety panel known as an institutional review board..monitored the testing of a vaccine its creators say prevents herpes outbreaks. Most of the 20 participants were Americans with herpes who were flown to the island several times to be vaccinated, according to Rational Vaccines...“What they’re doing is patently unethical,” said Jonathan Zenilman, chief of Johns Hopkins Bayview Medical Center’s Infectious Diseases Division. “There’s a reason why researchers rely on these protections. People can die.”...The risks are real. Experimental trials with live viruses could lead to infection if not handled properly or produce side effects in those already infected...
- FDA calls for industry input on continuous manufacturing guidelines (biopharmadive.com)
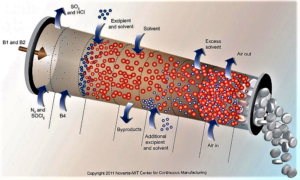
The Food and Drug Administration plans to develop clearer guidelines around the adoption of continuous manufacturing and has asked industry stakeholders for input on how best to design a regulatory framework...The regulator has advocated companies make the switch from traditional batch production to continuous manufacturing, which to a large extent hasn't been widely adopted by drugmakers despite the potential benefits to speed and reliability...continuous manufacturing can reduce the risk of manufacturing failures and potentially help prevent drug shortages from developing — areas that have been in focus as the agency steps up its oversight of pharmaceutical production facilities...Pharma has been loath to change over from batch production, which has served the industry for decades...Continuous manufacturing techniques — already adopted in many other industries — promise to shorten production times significantly, while reducing the amount of human intervention needed throughout...The FDA hopes providing a "framework of principles" will help drugmakers navigate CM adoption and implementation...
- First Gene-Transfer Therapy Approved for U.S. Market (ashp.org)Pioneering cancer drug, just approved, to cost $475,000 — and analysts say it’s a bargain (statnews.com)

FDA...announced the approval of tisagenlecleucel, a first-in-class chimeric antigen receptor (CAR) T-cell immunotherapy consisting of genetically modified autologous T cells, for the treatment of B-cell precursor acute lymphoblastic leukemia (ALL) in children and young adults...After modification in the laboratory and infusion back into the patient, the CAR T cells target and eliminate both normal and malignant CD19-expressing B cells. The genetic modification enhances the initiation of T-cell activation and the persistence of the transformed T cells...Novartis will market tisagenlecleucel as Kymriah. Labeling for the product states that it is indicated in patients up to age 25 years with ALL that is refractory or in second or later relapse...Tisagenlecleucel has an FDA-required risk evaluation and mitigation strategy (REMS) program that includes elements to assure safe use. The labeling states that the immunotherapy is available "only through a restricted program."...
- FDA Details Plans for More Efficient Inspections, Facility Evaluations

The US Food and Drug Administration's Center for Drug Evaluation and Research and Office of Regulatory Affairs will soon launch an effort to streamline the two offices' inspection and facility evaluation efforts...CDER Director Janet Woodcock and Associate Commissioner for Regulatory Affairs Melinda Plaisier said it is vital that the two offices quickly implement the plan in order to meet commitments under the recently reauthorized user fee agreements, specifically citing the agency's promise to communicate final inspection classifications to generic drugmakers within 90 days of an inspection beginning in October 2018...We plan to operationalize the plan in the fall of 2017 for nearly all human drugs...FDA details the plan—which includes specific operating models for pre- and post-approval inspections, surveillance inspections and for-cause inspections—in a 20-page white paper obtained by Focus entitled Integration of FDA Facility Evaluation and Inspection Program for Human Drugs: A Concept of Operations...