
- FDA oversight of the prescribing of fentanyl products is inadequate, report finds (healthcarefinancenews.com)Assessment of the FDA Risk Evaluation and Mitigation Strategy for Transmucosal Immediate-Release Fentanyl Products (jamanetwork.com)

The Food and Drug Administration and manufacturers did not take action when evidence emerged that potentially lethal fentanyl products were being inappropriately prescribed to patients, new research...shows...even as evidence emerged that as many as half of patients were taking dangerous medications known as TIRFs that should never have been prescribed to them, the FDA and fentanyl makers did not review prescribing records of even a single physician to consider disqualifying them from the program, which would have prevented them from prescribing the products...The study focused on Transmucosal Immediate-Release Fentanyls, or TIRFs, which are more dangerous than most prescription opioids on the market due to their very high potency and rapid onset. TIRFs are designed to get into the bloodstream within seconds, and because of their risks, were approved by FDA only for adult cancer patients "who are already receiving and who are tolerant to opioid therapy for their underlying persistent cancer pain."
- FDA plan would ease regulations for prescription drug apps (biopharmadive.com)

The Food and Drug Administration is seeking public comment on a proposed framework for regulating software applications developed by drugmakers for use in conjunction with their prescription drug products...The new approach would treat most prescription drug apps, including dose calculators, symptom trackers and medication reminders, as promotional labeling...drugmakers would need only to submit to the agency copies of the content of what the apps display to consumers, following existing reporting requirements for promotional materials...In other cases, such as when a drugmaker wants to show that software has an effect on a clinical outcome and wants to include information about the software in the FDA-required drug labeling, prior FDA approval would be required...
- Pharma’s slow embrace of continuous manufacturing (biopharmadive.com)

Widespread in other industries, continuous manufacturing features what's essentially an end-to-end assembly line, through which raw materials are steadily fed and constructed into final products...It's seen as faster and more flexible than the tried-and-true system of batching that forms the foundation of pharma manufacturing. The Food and Drug Administration is eagerly encouraging its use ...on a whole, the industry remains wedded to batch production. The reasons for why are many, but foremost is a reluctance to overhaul finely tuned manufacturing networks or to introduce new risks into drug development...Doing something different like continuous was going outside the box...Using an uninterrupted production process eliminates, or significantly reduces, the "hold times" in between steps that are typical in batch manufacturing. For J&J, production of Prezista used to take about two weeks from start to finish using batch methods...through continuous manufacturing, production takes only three days...since the raw ingredients don't need to transition in and out of production for quality testing, continuous manufacturing systems can run over a longer period of time, potentially upping output...For all of the advantages of continuous manufacturing over batch production, drugmakers must weigh switching over against retooling carefully crafted production networks.
- FDA’s continuing use of ‘black box’ for antidepressants ignores the harms of this warning (statnews.com)

The Food and Drug Administration’s “black box” warnings and advisories give important safety information about drugs. But they can sometimes go too far and harm more people than they help...Take the FDA’s highly publicized warnings that taking antidepressants increases the risk of suicidality...among children, adolescents, and young adults. We have evidence, as do many others, that these warnings have decreased youths’ access to mental health care and increased suicide attempts...In October 2004, the FDA required a so-called “black box warning” of this risk to be printed on the labels of all antidepressant drugs. It was implemented in January 2005. Two years later, the FDA extended the same warning to include young adults, again based on industry studies...There’s no question that antidepressants can cause harm if used inappropriately. But the FDA failed to recognize — and still won’t acknowledge — that the harms of its public advisories and black box warning on antidepressants for young people more than outweigh the benefits...
- FDA panel backs prescribing opioid overdose reversal drug along with painkillers (reuters.com)

An advisory panel to the Food and Drug Administration....recommended prescribing the opioid overdose reversal drug, naloxone, along with addictive painkillers...The recommendation of the panel underscores concerns about the growing opioid overdose epidemic...FDA studies found that co-prescribing naloxone to all patients who are prescribed painkillers could increase annual healthcare costs by $63.9 billion to $580.8 billion...I think co-prescribing is an expensive way to saturate the population with naloxone. The at-risk population is not necessarily the ones that are being prescribed new narcotics...
- FDA unveils open source code for collecting patient data (healthcareitnews.com)FDA’s MyStudies Application (App) (fda.gov)
The Food and Drug Administration launched a new app Wednesday to gather data for clinical trials and other research directly from patients...The FDA released the MyStudies app source code to the public, allowing developers and researchers to tailor the app to suit their research needs. The agency designed the app to facilitate the use of real-world data in research...Patients can submit real-world data to the app via their mobile devices. Researchers can then link those data to other electronic health information. The goal is to improve drug development...“Better capture of real world data, collected from a variety of sources, has the potential to make our new drug development process more efficient, improve safety and help lower the cost of product development,” FDA Commissioner Dr. Scott Gottlieb said in a statement. “Our hope is that the collection of more real world data directly from patients, using a secure app, will lead to more efficient product development and assist with safety monitoring.”
- The high cost of contamination in drugs manufacturing (pharmaceutical-technology.com)

Pharmaceuticals is one of the highest value industries globally..Any contamination in the drugs manufacturing process can have a substantial financial impact, not to mention possible safety implications...Drug manufacturer Genzyme was forced to temporarily close its Massachusetts plant in 2009, halting production of the Fabrazyme and Cerezyme drugs that were at the time used by 8,000 patients globally, after a virus was found to have contaminated a bioreactor...At the time it was estimated that the incident could cost Genzyme up to $300m in lost revenue...The company was later fined $175m by the US Food and Drug Administration and wrote off more than $28.4m worth of product...
- FDA keeps spotlight on GMP data integrity (biopharmadive.com)
The Food and Drug Administration...issued a reminder to drug manufacturers that data integrity remains high on the regulator's agenda of Good Manufacturing Practice-related concerns, publishing new guidance aimed at helping drugmakers meet its standards...The document, which updates a 2016 version, lays out a series of questions and answers for drugmakers explaining how companies can ensure manufacturing data sets are complete, consistent and accurate. While a technical concern, the FDA makes clear that data errors carry real risk to patient health...Guidance isn't the only lever the FDA can pull to help companies comply. In recent years, the agency has flagged data integrity issues in numerous warning letters following inspections and pre-approval assessments.
- Despite criticism and concerns, FDA approves a new opioid 10 times more powerful than fentanyl (statnews.com)Statement from FDA Commissioner Scott Gottlieb, M.D., on agency’s approval of Dsuvia and the FDA’s future consideration of new opioids (fda.gov)
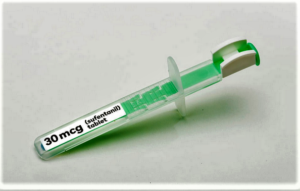
...the Food and Drug Administration approved an especially powerful opioid painkiller despite criticism that the medicine could be a “danger” to public health. And in doing so, the agency addressed wider regulatory thinking for endorsing such a medicine amid nationwide angst about overdoses and deaths attributed to opioids...The drug is called Dsuvia (sufentanil), which is a tablet version of an opioid marketed for intravenous delivery, but is administered under the tongue using a specially developed, single-dose applicator. These “unique features” make the medicine well-suited for the military and therefore was a priority for the Pentagon, a point that factored heavily into the decision…In discussing the rationale for the approval, however, Gottlieb argued that the different formulation and battlefield needs made it possible for the FDA to have Dsuvia fit into the “overall drug armamentarium.” And while he acknowledged the criticism, he insisted the risk management program, known as a REMS, will ensure the drug is only used in a medically supervised settings...AcelRx...worked with the Defense Department to develop Dsuvia...
- The search for new drugs is coming to your house (fastcompany.com)

Virtual trials could address some of the problems that come with developing new drugs, but a host of challenges remain before they can become routine...Last October, AOBiome Therapeutics...announced the results of a 12-week clinical trial of an experimental acne drug. In the randomized double-blind and placebo-controlled trial, a topical spray containing a beneficial bacteria was shown to be safe and effective in reducing the severity and number of acne lesions, the company said...The drug must successfully go through another round of testing–what’s called a Phase III trial–before AOBiome can apply to the Food and Drug Administration for marketing approval...Volunteers for the completed trial were recruited through social media and internet advertisements, and more than 8,000 people were screened online to see if they were eligible. The resulting 372 participants received the drug or a placebo in the mail and used company-issued iPhones to take selfies of their acne, a phone app to send the photos to physician-investigators for evaluation, and video conferencing to communicate with study staff...That’s markedly different from a typical drug trial...AOBiome’s acne drug trial, there were no in-person screening interviews, and no doctor visits. The trial was entirely what people in the trade call “virtual.”...Whether virtual clinical trials are the way forward is a matter of some debate, and industry analysts say a host of challenges remain before virtual trials–which currently represent a tiny fraction of the more than 100,000 registered clinical research studies in the United States–become the norm. These include overcoming a conservative corporate culture, ensuring that the technology is easy for patients to use, managing and analyzing the enormous amount of data that round-the-clock sensors generate, and proving the data’s reliability and validity to regulators...